
FDA Finalizes New National Drug Code Format
On March 5, 2026, the Food and Drug Administration (FDA) published a Final Rule adopting a new format for the National Drug Code (NDC). The rule takes effect on March 7, 2033. On the effective date, the FDA will assign new 12-digit NDCs and convert all previously assigned 10-digit NDCs to the uniform 12-digit NDC format.
What is the NDC?
The NDC is an FDA standard for uniquely identifying drugs marketed in the U.S. Currently, the NDC assigned by the FDA for each listed drug marketed in the U.S. is a unique 10-digit number and can be in several different formats.
Current formats:
10-digit identifier
The FDA’s standard NDC is a 10-digit numerical identifier that includes a labeler code, product code, and package code.
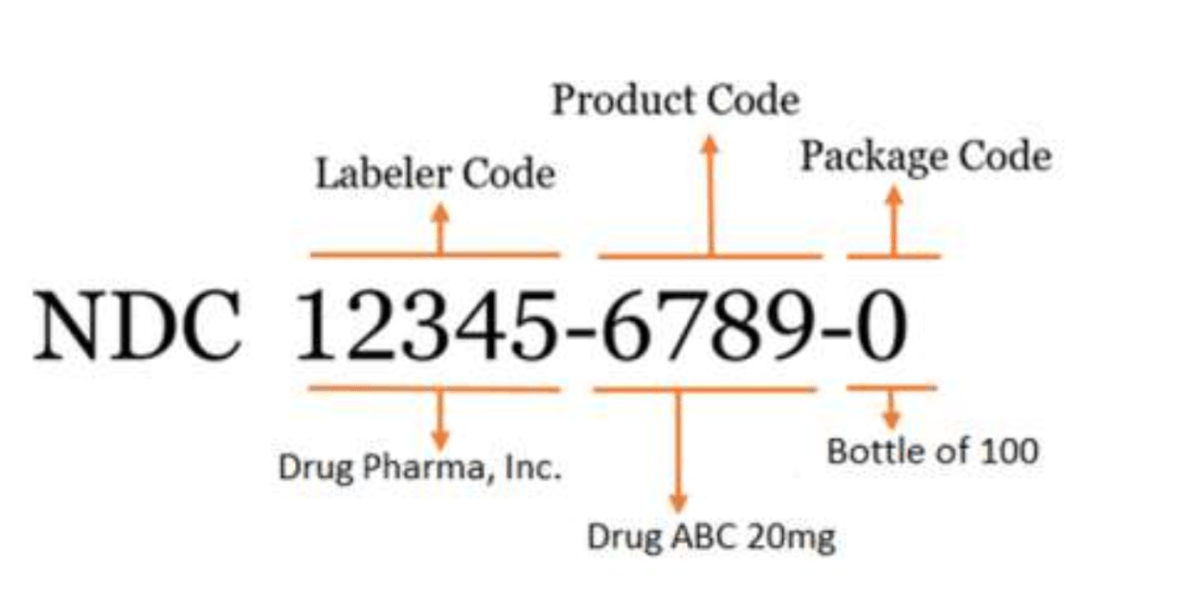
There are 3 FDA-assigned formats for the standard NDC:
- 4-4-2
- 5-3-2
- 5-4-1
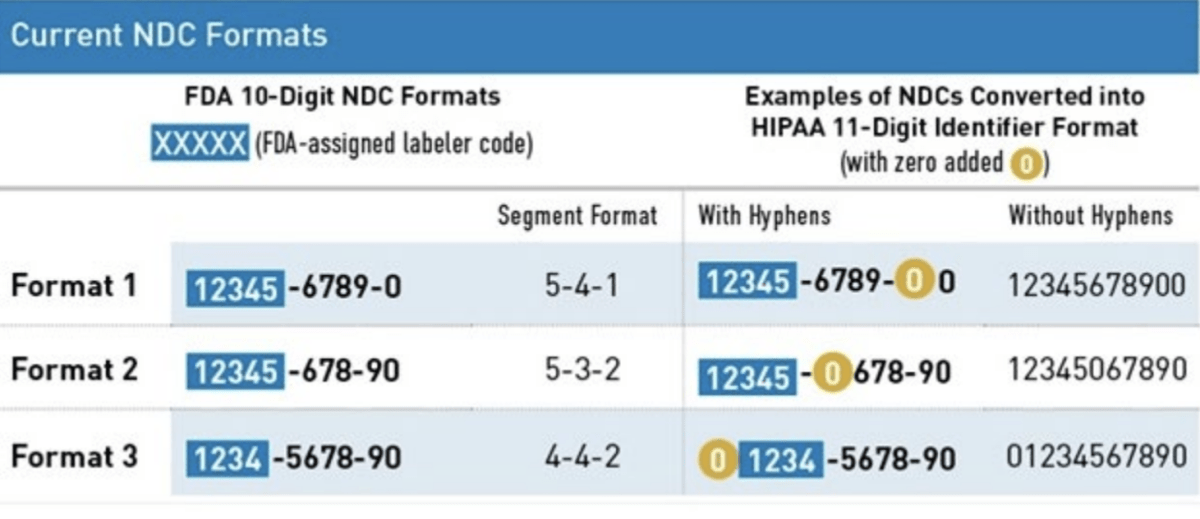
HIPAA Format
The Health Insurance Portability and Accountability Act (HIPAA) adopted a uniform 11-digit NDC format that must be used when a HIPAA-covered transaction includes an NDC. This 11-digit format is standardized into a 5-4-2 format and created by adding a leading zero to either the labeler, product, or package code.
Upcoming 6-Digit Format
The FDA will run out of 5-digit labeler codes in 10-15 years. Per FDA regulations (21 CFR 207.33), once the FDA runs out of 5-digit labeler codes, it will start assigning 6-digit labeler codes. Without this proposed change, there would be five NDC formats, 3 in 10- […]

{kind=link}
{kind=link}